Case Study 4: CFTR
star
star
star
star
star
Posljednje ažuriranje about 5 years ago
7

1
1
1
1
1
1
1
Background:
Cystic fibrosis is a common genetic disease caused by a mutation in a gene called the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The CFTR gene is located on chromosome 7 and has the directions to create the CFTR protein. The CFTR protein is a channel protein that regulates
how salts, most commonly sodium (Na+) and chloride (Cl-), and water move through the cell membranes of epithelial cells. Epithelial cells cover the surfaces of the body and can be found in the skin, respiratory, and digestive tracts.
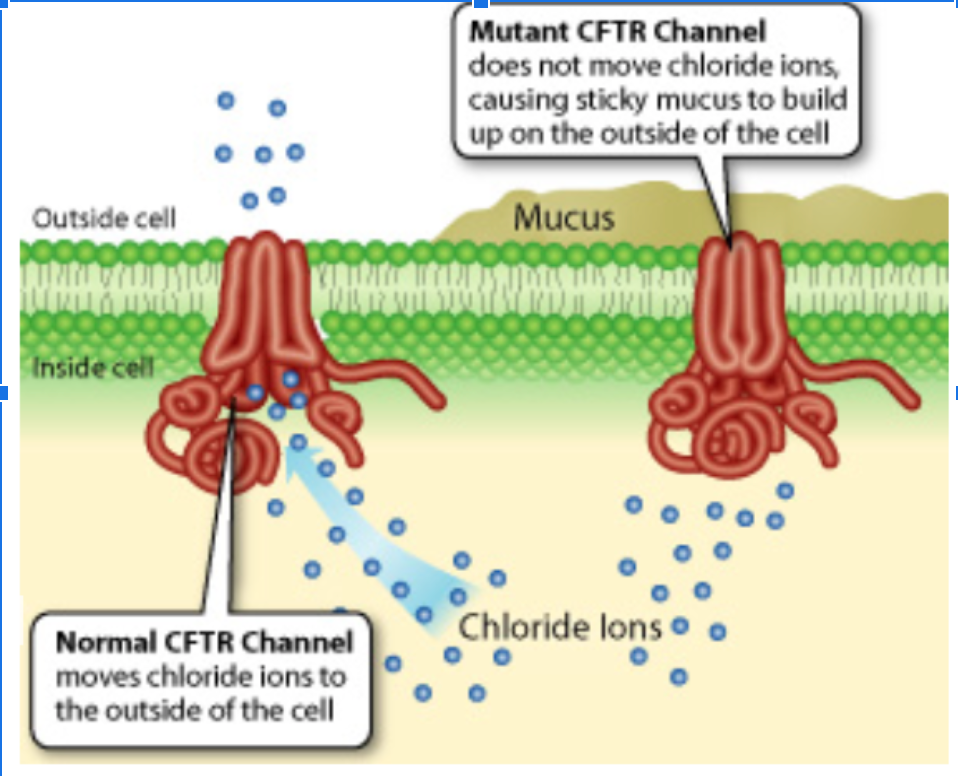
Na+ and Cl- help control the movement of water into tissues. When the CFTR protein does not function correctly, chloride (Cl-) is unable to pass through the center channel and sodium (Na+) is also unable to pass through the cell membrane. When they are imbalanced, watery substances like mucus are unable to move into the tissues and the mucus becomes extremely sticky and thick. (The role of mucus is to lubricate the surfaces of the body.) As a result, symptoms of cystic fibrosis include:
· Extremely salty skin
· Thick, sticky mucus that can block respiratory and digestive tracts
· Frequent respiratory infections due to bacteria trapped in mucus
· Wheezing, persistent cough, and shortness of breath
· Lack of digestion leading to poor growth/weight
The cystic fibrosis mutation is a recessive disorder passed from parent to offspring. This means an individual needs two copies of the mutated CFTR gene to have cystic fibrosis. Since the mutation is recessive, a parent may not have symptoms of cystic fibrosis or know they carry the mutation. There are more than 30,000 people in the U.S. with cystic fibrosis and more than 1,000 cases are diagnosed yearly. More than 10 million people in the U.S. are carriers of cystic fibrosis. When cystic fibrosis was first discovered, few sufferers lived past 6 years old, but due to medical advances the median age of survival has increased to 37 years old. With the new medication that just came out this year, it could be much longer!
What 3 bases in the gene were deleted during DNA replication to create the mutation? Provide evidence from Fig 1!
How do the codons differ between the normal and mutated mRNA transcripts? Provide evidence from Fig 1!
How do the amino acids differ between the normal and mutated Amino Acid sequences? Provide evidence from Fig 1!
Does a change in protein structure (amino acid order) cause a change in protein shape? Make a claim AND provide evidence for your claim- using evidence from figure 1 (AA sequence) and figure 2.
Does the mutation in CFTR genes result in a change in protein function? Make a claim AND provide evidence for your claim using figure 2.
Does the function of a persons' epithelial cells/tissues change if the function of the CFTR channel protein if a mutated?
Use evidence from the background information (short snip-ets or quotes) in your response.
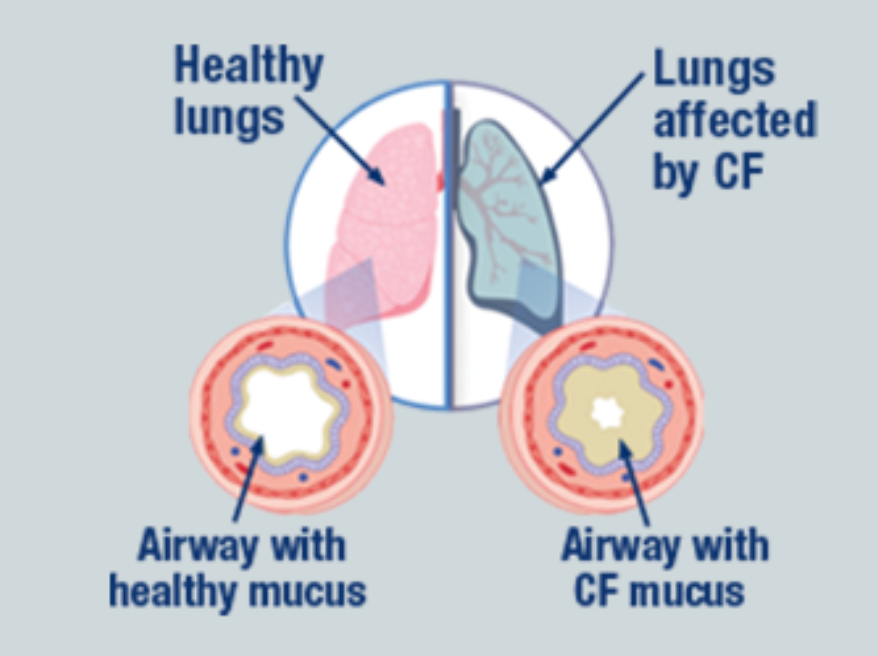
Does the function of the lungs and the overall feeling of the human, change if the function of the CFTR channel proteins change?
Use evidence from fig 3 and the background information to support your response.